分子が構築できましたので、計算を行う準備を始めます。
SetupメニューよりCalculationsを選択すると、下に示したCalculationsウィンドウが開きます。
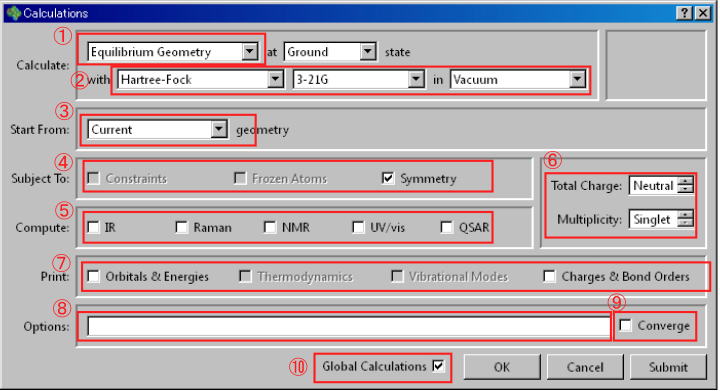
以下主だった設定について説明します。
Calculation部①
デフォルトを太文字にしてあります。
Energy 入力座標から構造を変化させずに、分子の持つ立体エネルギーを算出。 Equilibrium Energy 構造を最適化後、分子の持つ立体エネルギーを算出。 Transition Geometry 遷移状態の構造を求め、立体エネルギーを算出。 Conformer Distribution 配座解析を行い、安定配座のリストを作成。 Energy Profile 設定に従い段階的に構造を変化させて、それらの構造やエネルギーを求めリストを作成。
Transition Geometryはab initioや半経験的分子軌道方計算を選択しなければ計算する事が出来ません。
Calculation部② (選択した計算方法によってウィンドウの様子が変わります。
デフォルトを太文字にしてあります。
Molecular Mechanics - MMFF, MMFFaq, SYBYL - Semi-Empirical - AM1, RM1, PM3, PM6, MINDO - Hartee-Fock - STO-3G, 3-21G, 6-31G*, 6-31G**, 6-31+G*, 6-3111G, 6-311+G** Vacuum, Water, Acetone, Diethyl Ether, DMF, DMSO, Ethanol, Methylene Chloride, Toluene Density Functional BP, BLYP, EDF1, EDF2, B3LYP, M06、ωB97X-D 6-31G+, 6-31G**, 6-31+G*, 6-3111G, 6-311+G**, 6-311G(2df,2p), cc^pVTZ Moller Plesset MP2, RI-MP2 -
求めたい内容によって計算手法を選びます。
Start Form③
Start Formでは、計算を開始する,配座を選びます。通常はパソコン画面上の配座” Current,(デフォルト)"から計算しますが、コストの高い計算を行うには、コストの低い計算方法で安定配座を求めてから、最適化することにより、実際の計算での繰り返しを少なくして計算時間を短縮することができます。計算方法は
MMFF, AM1, PM3, 3-31G, 6-31G*, MMFF Conformer, MMFFaq conformer
から選ぶことができます。
実際には、Current以外を選択するのではなく、ファイル名を付けて、コストの低い計算方法で暫定的な安定配座を求めてから、計算結果を再最適化した方が、計算の手法が残せる。不必要に計算が長くなった場合ストップさせる等の自由度が高いように感じます。
Subject to ④
Constrain いくつかの結合パラメータを固定している場合にチェック。忘れると、設定したパラメータも最適化されてしまう為注意。
Energy Profileを選択した場合自動的にチェックされる。Frozen Atom 一つ以上の原子を固定するよう設定した場合にチェック。固定した原子は計算の過程で動きません。 Symmetry 対称性を持つ分子における計算を簡略化します。デフォルトでチェックが入っています。ほとんどの場合、そのままでいいはずです。
Copute⑤
計算したい内容を追加します。計算方法により選択できない場合もあります。スペクトルを求めるにはHartree Fock以上のレベルが必要です。Spartanか発者のHehre氏によると
NMR DFT B3LYP 6-31G*
UV/Vis DFT B3LYP 6-31+G*
レベルの計算が必要だそうです。
Charge, Multiplicity⑥
必要に応じて変更します。
個人的には電荷をもった分子の場合、どこかに存在する対イオンの位置によって構造は大きく変化してしまうことから、どのように使って良いのかよくわかりません。溶媒を選択することがよいのでしょうが、おそらく計算時間が増大します。
分子力場法では、電荷をもった官能基を想定してパラメータが作成されているか不安です。計算の信頼性を考える場合、半経験分子軌道法以上のレベルで計算させる必要があるように思います。
Print⑦
チェックされた項目がoutpuに書き出されます。計算の内容には影響しません。
Option⑧
計算の詳細についてoptionにキーワードを加え設定することができます。キーワードによって収束条件など詳細に設定することができます。
詳しくはマニュアルを参考にしてください。
よく使用するもののみ以下に示しました。
詳しくは、マニュアル、もしくはWavefunctionのホームページを参考にしてください。(Spartan06のキーワードの説明(日本語))
キーワード 値 説明 v SearchMethod C 配座検索の手法を選択します:初期設定では系の複雑さによって自動的にいずれかの手法を選択。
設定する場合、以下の様に入力する。
"SearchMethod=Systematic" 強制的に系統的な手法を用いる。
"SearchMethod=MonteCarlo" 強制的にMonte-Carlo 法を用いる。MaxItr N Monte Carlo法を用いた配座探索の繰り返し数をNに設定。
初期値は検索に含まれる結合と環の数で自動的に決定される。。
設定する場合、以下の様に入力する。(たとえば100回とした場合)
"MaxItr=100"GeometryCycle N 構造最適化等で最適化サイクルの最大値をNに設定。
初期値は、こ構造の複雑さによって自動的に決定される。
実際の計算で、iteration回数以内に構造最適化が終了しない場合「fail]となるが、GeometryCycleを大きな数値に変更すれば、問題が解決する。
10000回まで最適化する場合、
"GeometryCycle=10000"Gradient N 平衡構造および遷移状態の構造最適化における最大勾配成分の収束判定条件をF (hartrees/bohr単位)に設定
初期設定は4.5x10-4
Converge⑨
チェックすると収束を助けるための手順が実行されます。
Convergeは分子力学計算には使用でき無いそうです。
デフォルトはチェックされていませんが、変更する必要はほとんどないようです。
Global Calculation⑩
ファイル内に複数分子がと王禄されている場合に使用します。
チェックした場合 リストにある全ての分子を計算
チェックしない場合 画面上の分子のみを計算します。
リストへ分子の追加には、Fileメニューから “New Molecule”によって行い、リストの管理、変更チェックしない場合 はの作成は、Displayメニューの”Spreadsheet” で行います。